HANGİ GENETİK HASTALIKLAR İÇİN, HANGİ SOSYAL VE TEKNİK DURUMLARDA HANGİ BİYOPSİ VE PGT UYGULAMASI YÖNTEMİ YAPILMALIDIR?
Yrd. Doç. Dr. Hakan Berkil
Genetiks Genetik Hastalıklar Tanı Merkezi, Tıbbi Genetik Uzmanı, İstanbul
PGT Nedir?
Genetik bilimindeki son yıllardaki gelişmeler; tüp bebek yöntemiyle geliştirilen embriyolarda genetik incelemeler yapılmasına imkân tanımaktadır. Bu yönteme “embriyoda genetik tanı” (Preimplantasyon Genetik Tanı - PGT) adı verilmektedir. Gebelik öncesi genetik tanı adı da verilen PGT işlemi; yumurta ve
sperm hücrelerinin laboratuvar ortamında döllenmesi sonucunda gelişen embriyolardan 3. veya 5. günde alınan örneklerde gerçekleştirilmektedir.
Alınan hücrelerde özel yöntemler kullanılmakta ve sayısal ve yapısal kromozom bozuklukları ile tek gen hastalıklarının (Akdeniz Anemisi, Orak hücreli Anemisi, Spinal Musküler Atrofi (SMA), Kistik Fibrozis gibi) tanısı yapılabilmektedir.
PGT’nin Amacı Nedir?
Bireylerin; taşıdıkları kalıtsal hastalığı değişik oranlarda çocuklarına aktarma riskleri nedeniyle genetik hastalıkların bireylerde ve embriyolarda belirlenmesi çiftlerin sağlıklı çocuk sahibi olabilmesi için önemlidir. Günümüzde; farklı teknikler kullanılarak, birçok kalıtsal hastalığın tüp bebek aşamasında embriyolar ana rahmine konmadan önce tanımlanması mümkün hale gelmiştir.
PGT’nin asıl amacı; ailede daha önce saptanmış kromozom ve DNA hastalıklarının embriyo aşamasında tanımlanmasıdır. Ayrıca, infertilite nedeni ile tüp bebek tekniklerinin uygulanacağı çiftlerde embriyolarda oluşması muhtemel genetik bozukların tanımlanması için de kullanılmaktadır.
PGT’nin Avantajları Nelerdir?
• Ailelerin sağlıklı çocuk sahibi olmaları sağlanmaktadır.
• Gebelik kayıpları azaltılmaktadır.
• Ailenin gebe kalma şansı arttırılmaktadır.
• Aile, gebelik sonlandırılmasına bağlı tıbbi ve psikolojik sorunlardan korunmaktadır.
• Talasemi vb. hastalıklarda doku tiplemesi ile hasta çocuk için tedavi imkânı sağlanmaktadır.
• PGT; gebelik kayıpları nedeniyle ailelerin yaşadığı sıkıntılar ve doğan hasta çocukların yaşam boyu karşılaştıkları sağlık problemleri, hastalıkların tedavisindeki güçlükler ve yüksek tedavi maliyetleri ile karşılaştırıldığında çok daha faydalı ve ucuz bir tanı yöntemidir.
PGT’de Kullanılan Yöntemler Nelerdir?
Çiftin tüp bebek tedavisine alınmasından sonra yaklaşık 10-12 günlük tedavinin ardından yumurta hücreleri toplanarak her biri ayrı bir sperm hücresi ile döllenir. Döllenen ve iyi gelişen (embriyo) 3. günde blastomer biyopsisi ile 1 adet hücre veya 5. günde trofoektoderm biyopsisi ile 4-5 tane hücre alınır. Daha sonra; floresan in situ hibridizasyon (FISH), array komperatif genomik hibridizasyon (aCGH), Yeni Nesil Dizileme (NGS), PCR, DNA dizi analizi ve fragman analizi adı verilen farklı tekniklerden bir veya birkaçı kullanılarak bu hücrelerde genetik inceleme yapılır (Tablo 1).

PGT ile Hangi Hastalıklar Tanımlanabilmektedir?
Günümüzde, bazı özel durumlar dışında hemen hemen tüm genetik hastalıklarda PGT uygulanabilmektedir. PGT işleminin uygulanabilmesi için en önemli koşul ise ailede bulunan genetik hastalığın tanımlanmış olmasıdır. Aksi durumda, PGT uygulanamaz.

PREİMPLANTASYON GENETİK TANI (PGT) ENDİKASYONLARI Ve UYGULAMA İÇİN GEREKLİ KOŞULLAR
PGT, genetik bir hastalığı olan veya kalıtsal bir hastalık için taşıyıcılık saptanmış ve sağlıklı çocuk sahibi olmak isteyen çiftlere önerilmektedir. Özellikle, hastalık riski yüksek olduğu için tek gen hastalıkları veya kromozom bozukluğu saptanmış çiftlerin çocuklarında günümüzde sıkça uygulanmaktadır. Benzer olarak, ailesel kanser tablosuna neden olan genetik değişikliklerin saptandığı çiftlerde de kullanılabilmektedir. Ayrıca, kendilerinde bir genetik problem bulunmamasına rağmen infertilite nedeniyle tüp bebek tekniklerinin uygulanacağı ailelerde de PGT önerilmektedir. Özellikle; ileri anne yaşı (37 yaş ve üstü), tekrarlayan gebelik kayıpları ve tekrarlayan tüp bebek denemelerinde başarısızlık bulunan çiftlerin gebeliklerinde kromozom hastalıklarının görülme riski yüksek olduğu için PGT uygulanmaktadır.

KROMOZOMAL HASTALIKLAR
YAPISAL KROMOZOM BOZUKLUKLARI
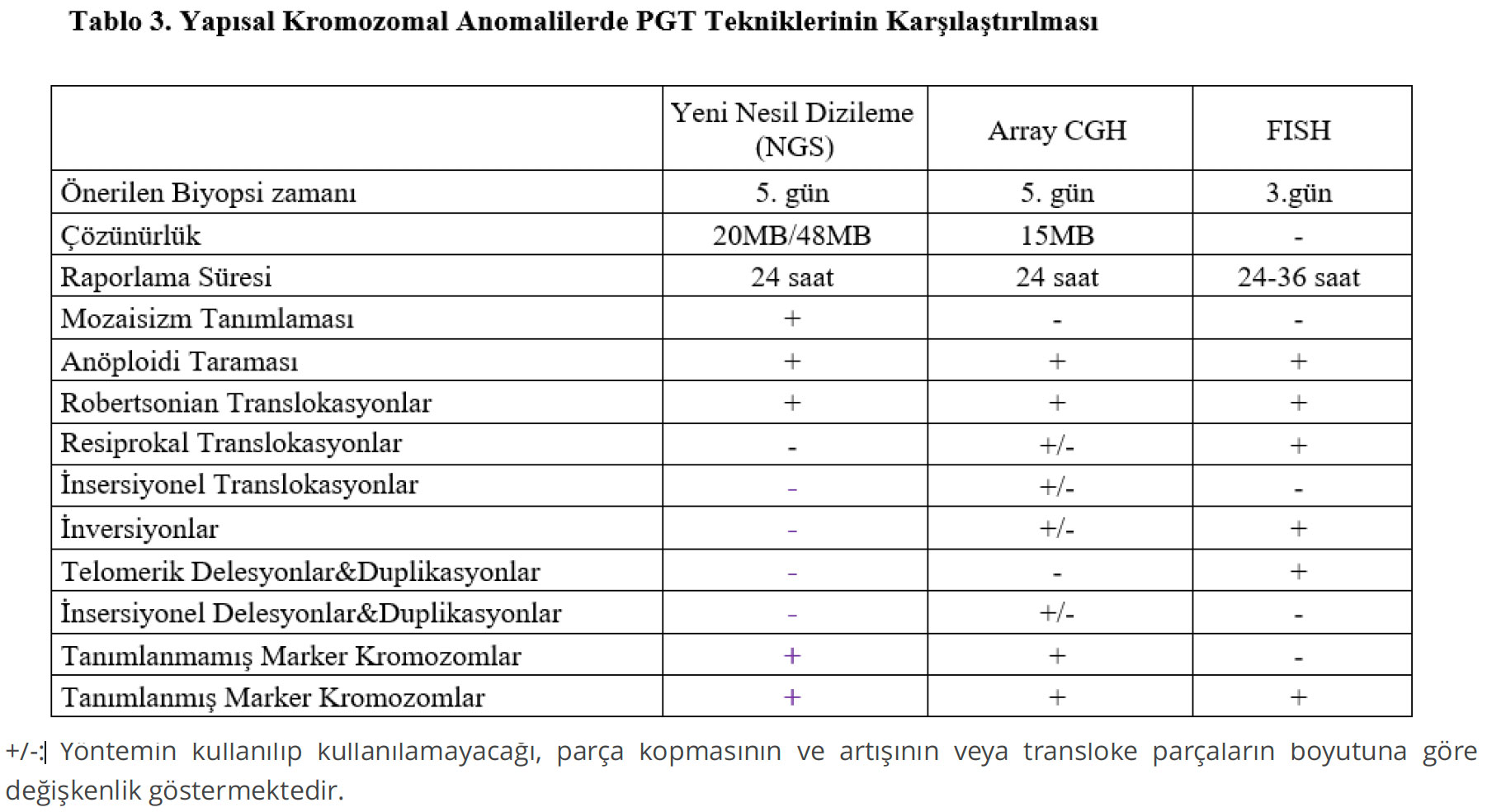
Kromozom analizlerinde translokasyon ve inversiyon gibi yapısal kromozom bozukluğu saptanan çiftlerin embriyolarında dengesiz kromozom yapısı görülme riski ciddi oranlarda artmaktadır. Bu nedenle, yapısal kromozomal bozukluk saptanan çiftlere bu konuda gerekli genetik danışma verilmeli ve olası riskler anlatılarak PGT (PGT-SR) önerilmelidir. Marker kromozom taşıyıcısı bireylerde, marker kromozomun köken aldığı kromozomun monozomileri ve trizomileri gözlenebileceği için aCGH veya NGS yöntemlerinden birisi ile PGT-SR uygulanmalıdır.
Günümüzde kullanılan mevcut tekniklerden birisi ile saptanabilir olması durumunda, daha nadir gözlenen yapısal kromozomal bozuklukları içerisinde yer alan insersiyonel translokasyon veya hafif klinik bulgular ile seyreden kromozomal delesyon (parça kopması) veya duplikasyonlara (parça artışı) sahip bireylerin saptandığı çiftlere PGT-SR önerilmelidir.
Yapısal kromozom bozukluğuna sahip çiftlerde, hazırlanan özel problarla FISH veya uygun olması durumunda 24 kromozom inceleme olarak da adlandırılan aCGH ve NGS tekniklerinden birisi kullanılarak PGT-SR yapılarak sağlıklı embriyolar tanımlanabilir. Bu sayede, tekrarlayan gebelik kayıpları ve hasta çocuk doğumları önlenebilir.
Bazı Robertsonian translokasyon tiplerinde, ailenin YÜT + PGT-SR uygulansa dahi çiftlerin sağlıklı çocuk sahibi olma şansı bulunmamaktadır. Robertsonian translokasyonun bir türü olan ve akrosentrik homolog kromozomların translokasyonlarını taşıyan çiftlerde oluşacak tüm embriyolar ilgili kromozom için monozomik veya trizomik yapıda olacaktır. Bu nedenle; saptanan translokasyon için mozaik yapı saptanmadığı sürece, aşağıda belirtilmiş olan kromozom yapılarından birisine sahip çiftlerde YÜT + PGT-SR uygulanmamalıdır.
• 45,--, rob(13;13) veya 45,--,t(13;13)
• 45,--, rob(14;14) veya 45,--,t(14;14)
• 45,--, rob(15;15) veya 45,--,t(15;15)
• 45,--, rob(21;21) veya 45,--,t(21;21)
• 45,--, rob(22;22) veya 45,--,t(22;22)
Yine, sıklıkla raporlanan 1qh+, 9qh+, 16qh+, Yqh- gibi heterokromatin bölge artış ve azalmaları ile sadece akrosentrik kromozomlarda bulunan satellit yapısındaki artışlar (21ps+ vb.) ve inv(9)p12q13) “An International System for Human Cytogenomic Nomenclature 2016” değerlendirmesine göre “normal kabul edilen kromozomal varyasyonlar” grubunda bulunmaları nedeniyle PGT-SR uygulanmasına gerek yoktur. Ayrıca; heterokromatin bölge değişiklikleri ve satellit artışları kromozomal hastalık olarak kabul edilseler dahi bugünkü teknikler ile PGT uygulaması mümkün değildir.
PGT Uygulaması İçin Gerekli Koşullar
Yapısal kromozom bozukluğu saptanan çiftlerin embriyolarında FISH, aCGH, NGS gibi farklı PGT seçenekleri mevcut olup hasta için en uygun olan yöntemin seçilmesi doğru tanı açısından önemlidir. Bu nedenle, her olgunun saptanmış olan kromozomal hastalık tipine ve kromozomlardaki kırık noktalarına göre PGT öncesinde bir genetik uzmanı tarafından değerlendirilmesi ve en uygun tekniğin belirlenmesi gereklidir. Kromozom analizi raporunda belirtilen kırık bölgelerinin ve yer değiştiren kromozom parçalarının büyüklüklerinin değerlendirilerek aCGH ile saptanıp saptanamayacağının önceden kontrol edilmesi gereklidir. Translokasyon, inversiyon vb. yapısal kromozom bozukluklarında kırık noktalarından birisinin telomerik bölgede veya bu bölgeye yakın olması durumunda FISH tekniği ile PGT uygulaması zorunludur. aCGH tekniği telomer bölgelerini güvenli olarak analiz edemediği için, NGS yöntemi de bu hastalarda kullanım için üretici firmaları tarafından onaylanmadığı için kullanılmamalıdır.
Gerekli olan malzemelerin (FISH probları) mevcut olup olmadığının kontrol edilmesi ve kullanılacak prob birleşiminin belirlenebilmesi FISH ile PGT öncesinde daha YÜT uygulaması başlamadan genetik tanı merkezine mutlaka bilgi verilmelidir. Robertsonian translokasyon ve inversiyonlarda iki farklı prob kullanılması PGT-SR çalışması için yeterli olurken resiprokal translokasyonlarda 4 hareketli kromozomal parça olduğu olası kromozomal dengesizliklerin tanımlanabilmesi için en az üç farklı prob kullanılmalıdır. Bu durum, Robertsonian translokasyon ve inversiyonlarda 24 saat olan PGT süresini resiprokal translokasyonlarda 36 saate çıkarmaktadır.
FISH ile PGT-SR uygulamalarında PGT uygulaması, kadın doğum uzmanının kararına göre, 3. günde embriyolardan alınacak tek bir blastomer üzerinde yapılarak 24-36 saat içerisinde raporlanabilir. İstenirse, 5. günde alınan trofoektoderm biyopsi örneklerinde de FISH tekniği uygulanabilir. Ancak, sonuç süresi nedeniyle bu tür olgularda biyopsi sonrası embriyoların dondurulması gereklidir.
Kırık noktalarının telemore yakın olmadığı ve yer değiştiren parçaların 15MB ve üzerinde olduğu olgularda aCGH tekniği kullanılabilir. Aksi halde, hem telomerik bölgeler için net bilgi vermemesi, hem de çözünürlük olarak adlandırılan görme kapasitesinin altında kalması nedeniyle bu tekniğin kullanılması hatalı sonuçlara neden olabilir. Robertsonian translokasyonlarda, embriyolarda translokasyona katılan kromozomların anöploidileri (monozomi veya trizomi) gözleneceği için aCGH yöntemi güvenli olarak uygulanabilir. Ayrıca, kromozomlar arasındaki total kısa ve uzun kol değişimlerinin saptandığı kişilerde de aCGH kullanılabilir.
NGS kitleri, yeni nesil dizileme tekniğinin embriyolardaki anöploidilerin tanımlanması için geliştirilmiş kitler olup bu tür çiftlerde kullanılması önerilmemektedir. Bunun nedeni, çözünürlüklerinin aCGH tekniğine göre daha düşük olması ve üretici firmalar tarafından bu tür olgularda kullanılmasının onaylanmamasıdır. Günümüzde dünyada yoğun olarak kullanılan VeriSeqTM (illumina) ve ReproSeqTM (ThermoFisher) kitlerinin çözünürlükleri üretici firmalar tarafından sırasıyla 20MB ve 48MB olarak bildirilmiştir. Bu nedenle; çözünürlüklerinin altında kalan parça kopması ve artışları güvenilir olarak saptamaları teknik olarak mümkün değildir.
Marker kromozom saptanan çiftlerde, sıklıkla marker kromozomun kökeni tanımlanamasa da köken aldığı kromozomun anöploidilerine neden olabileceği için aCGH veya NGS tekniklerinden birisi ile PGT-SR uygulanmalıdır. İnsersiyonel translokasyona sahip kişilerde transloke olan parçanın boyutlarının 15MB veya daha büyük olduğu düşünülüyor ise aCGH tekniği ile PGT önerilebilir. Aileye, saptanmış olan transloke parçanın gerçek büyüklüğünün hasta bir çocuk olmadığı sürece kesin olarak belirlenemeyeceği, bu nedenle de hata payı olabileceği, ancak tek PGT seçeneğinin aCGH yöntemi olduğu ve bu şartlarda PGT uygulaması yapılması durumunda da mutlaka prenatal tanı seçeneklerinden birisi ile doğrulama yapılması gerektiği anlatılmalıdır.
Hafif klinik bulgular ile seyreden kromozomal parça kopmasına sahip kişilerde genellikle telomerik bölgelerde kayıplar saptanmaktadır. Bu tür çiftlerde ilgili kromozom için telomerik problar kullanılarak FISH tekniği ile PGT-SR uygulaması yapılabilir.
Özellikle; resiprokal translokasyonlu olgularda transfer edilebilecek embriyo bulmak daha zor olduğu ve PGT maliyetleri de yüksek olduğu için birden fazla YÜT uygulanarak embriyo biriktirme işlemi yapılması önerilmelidir. Ülkemizde, PGTSR ücretlendirmeleri 12-15 embriyoya kadar sabit bir fiyatlandırma üzerinden yapıldığı için PGT işleminin tek bir çalışmada bu sayılara yakın sayıda embriyoda gerçekleştirilmesi hem hastaların PGT maliyetini azaltmakta hem de transfer şansını arttırmaktadır.
aCGH ve NGS tekniklerinden bir tanesinin uygulanması durumunda çalışma 3. gün veya 5. gün biyopsi örneklerinden yapılabilir. Embriyo dondurma maliyetleri ve/veya taze embriyo transferi tercihleri nedeniyle farklı merkezler farklı biyopsi seçeneklerini kendileri için daha uygun bulmaktadır. Ancak, gerek mozaisizm gerekse daha güvenilir sonuçların elde edilebilmesi için 5. gün trofoektoderm biyopsi örnekleri tercih edilmelidir. Ayrıca, aCGH ve NGS analizlerinde çalışmanın ilk aşaması tüm genom çoğaltma (WGA) işlemi olup bu çalışmanın tek bir blastomer yerine trofoektoderm hücrelerinden yapılması elde edilen ürünün kalitesini arttırır ve analiz sonuçlarının çok daha güvenilir olmasını sağlar.
aCGH ve NGS çalışmaları embriyo başına ücretlendiği için 3. gün biyopsi örneklerinden çalışma yapılmasının hastaların PGT maliyetlerini arttırdığı unutulmamalıdır. Kontaminasyon riski, biyopsi örneklerinin yanlış numaralandırılması gibi karışıklıkların geriye dönük kontrolü gerektiğinde yapılabilmesi için WGA işleminin mutlaka embriyoloji laboratuvarından gönderilen tüplerde yapılması önerilir. aCGH ve NGS çalışmalarında, elde edilen WGA ürününün sadece bir kısmı kullanıldığı için herhangi bir şüpheli durumda kalan ürünlerden çalışma tekrar edilebilir.
SAYISAL KROMOZOM BOZUKLUKLARI
X ve Y kromozomlarında sayısal bozukluk saptanmış çiftlerde cinsiyet kromozomları ile ilgili hastalıkların görülme olasılığı yüksektir. Özellikle, Klinefelter (47,XXY), XXY, XXX ve X kromozom mozaisizmi saptanan kişilerin embriyolarında benzer veya farklı bir cinsiyet kromozom hastalıklarının oluşma riski artmaktadır.
Teknik imkânların gelişmesi ile birlikte uzun zamandır Klinefelter sendromlu hastalar da çocuk sahibi olabilmektedirler. Bu kişilerde yapılan cerrahi işlemler sonrasında sperm bulunması durumunda YÜT uygulanabilmektedir. Ancak, kişide saptanan sayısal kromozomal hastalık nedeniyle bazı embriyolarda kromozomal hastalıklar gözlenebilir. Bu embriyoların genetik inceleme yapılmadan anne adayına transfer edilmesi durumunda gebeliğin oluşmaması, biyokimyasal abortus ve gebelik kaybı gibi klinik tablolar ortaya çıkabilmektedir. Ayrıca, olası gebeliklerde Klinefelter hastalığına çocukların doğma ihtimali bulunmaktadır. Sıklıkla tesadüfen saptanan XXX ve XYY olgularında da embriyolarda XXY (Klinefelter) kromozom yapısı gözlenebileceği unutulmamalıdır.
PGT Uygulaması İçin Gerekli Koşullar
Sayısal kromozom bozukluğuna sahip çiftlerde standart FISH panellerinden veya aCGH ve NGS tekniklerinden birisi kullanılarak PGT uygulanabilir ve tekrarlayan gebelik kayıpları ile etkilenmiş çocuk doğumları engellenebilir. Sayısal kromozom bozuklukları, PGT uygulaması açısından özel bir değerlendirme gerektirmediği için kadın doğum uzmanı tarafından uygun görülen PGT-A (FISH, aCGH veya NGS) yöntemlerinden birisi seçilerek yapılabilir.
Kromozom Anomalili Çocuk Öyküsü
Kromozomal bozukluklar genellikle ölümcül olup canlı yeni doğanlarda görülme sıklığı %0,5-0,7 arasındadır. Sıklıkla gebelik sırasında, özellikle de ilk 3 ay içerisinde gebelik kayıplarına neden olurlar. Gebelik kayıplarına neden olan sayısal kromozomal bozuklukların tekrarlama riskleri, genellikle embriyoda yeni oluşmuş (de novo) düzensizlikler olmaları nedeniyle çok düşüktür. Ancak; bu tür kromozomal bozukluklar birden fazla gebelikte meydana gelebilmekte ve hasta çocuk (lar) doğabilmektedir. Kromozom yapıları sıklıkla normal saptanan bu çiftlerde, üreme hücreleri ile sınırlı kromozomal birhastalık taşıyıcılığının (germline mozaisizm) veya yumurta ve sperm hücrelerinin olgunlaşması sırasında meydana gelen segregasyon bozukluklarının tekrarlayan gebelik kayıplarına veya hasta çocuk doğumlarına yol açabileceği unutulmamalıdır.
PGT Uygulaması İçin Gerekli Koşullar
Bu tür çiftlerde, YÜT uygulanması durumunda kadın doğum uzmanı tarafından uygun görülen PGT-A (FISH, aCGH veya NGS) yöntemlerinden birisi seçilerek yapılabilir.
İnfertilite Nedeniyle YÜT Uygulanan Çiftler Kişilerde saptanan sayısal ve yapısal kromozom bozukluklarının dengesiz genetik yapıya sahip embriyoların oluşma riskini ciddi oranda arttırması ve YÜT maliyetleri nedeniyle infertilite nedeniyle başvuran çiftlere kromozom analizi mutlaka yapılmalıdır. Sayısal veya yapısal kromozomal bozukluk saptanan çiftlere bu konuda gerekli genetik danışma verilmeli ve olası riskler anlatılarak prenatal veya PGT önerilmelidir.
Genetik incelemeler sonrasında kromozomal bozukluk saptanmamış bazı çiftlerde de PGT önerilmektedir. Çiftlerde; anne yaşının 37 ve üzerinde olması, tekrarlayan gebelik kayıpları ve kaliteli embriyolar transfer edilmesine rağmen birden fazla YÜT denemesinde başarısızlık gibi durumlarda PGT-A işlemi yapılabilmektedir.
37 yaş ve üzeri kadınlarda, yumurta hücresinin olgunlaşma mekanizmasında ilerleyen yaş ile birlikte sorunlar ortaya çıkmaktadır. Bunun neticesinde; embriyoların ana rahmine tutunmaması (gebelik oluşmaması), gebelik oluşursa doğuma ulaşmadan kaybedilmesi veya kromozom bozukluğuna sahip çocuk doğması gibi sıkıntılar karşımıza çıkmaktadır.
Tekrarlayan düşükleri bulunan çiftlerde yapılan araştırmalarda, gebeliğin ilk 3 ayındaki tekrarlayan gebelik kayıplarının %50-60’da kromozomal bozukluklar saptanırken bu oran ikinci 3 ayda %20-25, son 3 ayda ise %5-10 olarak belirlenmiştir. Bu çiftlerin birçoğunda kromozom yapısı normal saptanmakla birlikte bu kişiler bazı kromozomal bozukluklar için sadece üreme hücrelerini kapsayan mozaik bir yapıya veya sağlıklı gerçekleşmeyen bir segregasyon mekanizmasına sahip olabilirler.
Ayrıca, birçok kez yardımcı üreme teknikleri kullanılmasına ve iyi kalitede embriyolar verilmesine rağmen gebelik sağlanamayan çiftlerin embriyolarında genetik bozuklukların varlığı söz konusu olabilmektedir. Şiddetli erkek infertilitesi saptanmış çiftlerin embriyolarında yapılan genetik çalışmalarda yüksek anöploidi oranları
saptanmıştır.
PGT Uygulaması İçin Gerekli Koşullar
Yukarıda belirtilen nedenlerden dolayı bu tür çiftlerde gerekli görülmesi durumunda kadın doğum uzmanı tarafından uygun görülen PGT-A (FISH, aCGH veya NGS) yöntemlerinden birisi seçilerek uygulanabilir.

Tek Gen Hastalıkları
Mendelyen kurallara göre kalıtılan tek gen hastalıkları kromozomlar üzerinde yer alan tek bir gendeki mutasyonlardan kaynaklanır. Bu değişimler sonucunda oluşan genetik hastalıkların kalıtım modeli, otozomal veya gonozomal kromozomlardaki genlerden köken almasına göre değişmektedir. Otozomal kalıtımlı hastalıklar, bir çift otozomal kromozom üzerinde bulunan aynı loküsteki tek bir genin veya her ikisinin mutasyona uğraması durumuna göre resesif Meme kanserli hastalarda kontrollü overyan stimülasyon nasıl yapılmalı? veya dominant olarak adlandırılmakta ve tekrarlama riskleri de farklılık göstermektedir. Birbirinden bağımsız birden fazla farklı gen bölgesi aynı hastalık tablosuna neden olabilir. Bu nedenle, mutant genin otozomal veya gonozomal kromozomlarda bulunmasına göre tekrarlama riskleri aynı hastalık için değişkenlik gösterebilir. Ayrıca çevresel faktörler ile oluşmuş hastalıkların bir kısmı genetik hastalıkları taklit edebilir ve tekrarlama riski olmamasına rağmen aileye yanlış bilgi verilmesine neden olabilir.
Günümüzde; DNA dizi analizi ve fragman analizi gibi yöntemlerle Talasemi, Kistik Fibrozis, Spinal Musküler Atrofi (SMA), Hemofili, Duchenne/Becker Musküler Distrofi vb. birçok genetik hastalık embriyo düzeyinde tanımlanabilmektedir. Bu yöntemle; aile, gebelik döneminde bebekte genetik bir hastalık ortaya çıkması nedeniyle uygulanan gebelik sonlandırılmasına bağlı tıbbi ve psikolojik sorunlardan korunmaktadır. Ayrıca; hasta kişilerin yaşam boyu karşılaştıkları sağlık problemleri, hastalıkların tedavisindeki güçlükler ve yüksek tedavi maliyetleri nedeniyle de çok önemlidir.
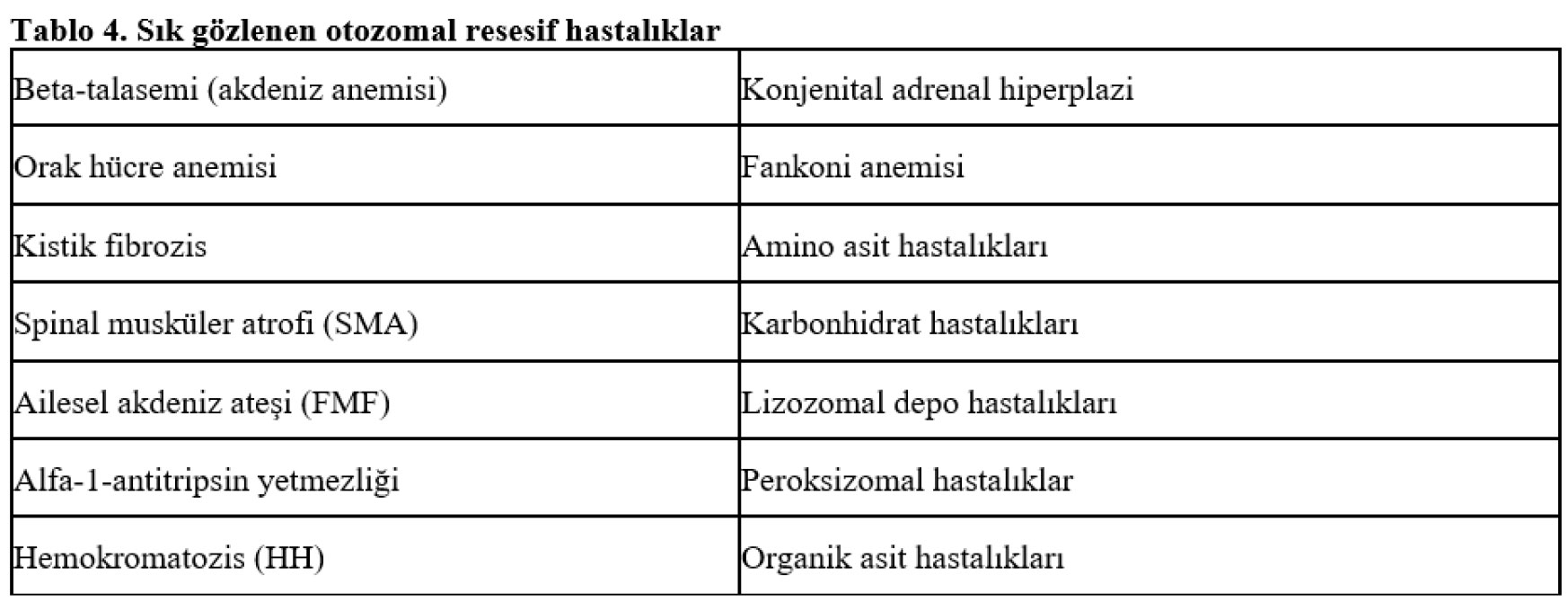
Otozomal Resesif Hastalıklar (OR)
Otozomal resesif hastalıklar tek gen hastalıkların yaklaşık üçte birini oluştururlar. Hastalığın ortaya çıkabilmesi için aynı loküste yer alan bir gen çiftinin her ikisinde de fonksiyonel değişikliğin oluşmasının gerekli olduğu hastalıklar otozomal resesif kalıtımlı hastalıklar olarak adlandırılırlar. Bu hastalıklar her kuşakta kendini göstermez ve aile ağacı yatay özellik gösterir. Ebeveynler genellikle sağlıklıdır ve hastalık aynı kuşakta hasta kişinin kardeşlerinde gözlenir. Akraba evlilikleri, kişilerin ortak bir atadan gelmelerine bağlı olarak aynı mutant geni taşıma risklerinin artması nedeniyle otozomal resesif hastalıkların görülme sıklığını arttırır.
Hastalık cinsiyet ayırımı göstermez ve aynı şiddette seyreder. Taşıyıcı çiftlerde her gebelik için hastalığın ortaya çıkma riski %25’tir. Resesif hastalıklarda çocukların taşıyıcı olma riski %50 iken çocuklarda mutant genin taşınmama şansı %25’tir. Taşıyıcı olan kişi, normal yani mutant geni taşımayan bir kişi ile evlendiğinde çocuklarında hastalığın görülme riski yoktur. Hasta bir kişinin mutant geni taşımayan bir kişi ile evlenmesi durumunda ise çocukların tümü sağlıklı fakat
taşıyıcı olacaktır. Hasta bir kişinin taşıyıcı bir birey ile evlenmesi durumunda ise her gebelik için çocukların hasta olma riski %50 iken taşıyıcı olma riski de % 50’dir.
Talasemi gibi bazı hastalıklar için toplumumuzdaki taşıyıcılık frekansının yüksek olması ve akraba evliliklerinin sık yapılması nedeniyle ülkemizde otozomal resesif hastalıklar daha sık gözlenmektedir. Bu nedenle; özellikle Akdeniz Anemisi, Orak Hücre Anemisi, Spinal Musküler Atrofi (SMA) ve Kistik Fibrozis hastalığına sahip çocukları olan ailelere veya taşıyıcı oldukları önceden saptanan çiftlere PGT-M önerilmelidir.
Otozomal Dominant Hastalıklar (OD)
Aynı loküste yer alan bir gen çiftinin sadece bir tanesinde oluşan fonksiyonel değişikliğin hastalığın oluşması için yeterli olduğu hastalıklar otozomal dominant kalıtım örneği gösterirler. Bu hastalıklarda sıklıkla normal gen ürünü yapısal bir proteindir. Aile ağacı dikey özellik gösterir. Genetik hastalık, her kuşakta kendini gösterir ve hastalığın saptandığı kişinin (indeks olgu) etkilenmiş bir ebeveyni bulunur. Otozomal dominant kalıtılan hastalıklarda etkilenmiş kişinin çocuklarında hastalığın ortaya çıkma riski hasta kişinin ve çocuklarının cinsiyetinden bağımsız olarak her gebelikte %50’dir. Yukarıda bahsedilen klasik kalıtım modelinin aksine otozomal dominant hastalıkların çoğu yeni mutasyonlardan kaynaklanır ve bu nedenle de etkilenmiş bir ebeveyn saptanmaz. Ayrıca, dominant hastalıklarda hastalığın aktarıldığı kişide fenotipik bulguların şiddeti değişiklik gösterebilir (ekspresyon) veya mutant genin aktarıldığı kişide hastalık ortaya çıkmayabilir (penetrans kaybı). Bu nedenle hastalığın her kuşakta kendini göstermesi ve dikey aile ağacı özelliği gözlenmez. Diğer istisnai durumlardan birisi de, hastalığın sadece ebeveynlerden birinin gamet hücrelerindeki
mutasyonlardan kaynaklanmasıdır. Bu durumda, yeni bir mutasyon nedeniyle oluştuğu düşünülen ve bu nedenle de tekrarlama riski olmadığı kabul edilen bir hastalıkta risk bu mutasyonun üreme hücrelerinde taşınma oranına göre değişiklik gösterir.
Nörofibromatozis, polikistik böbrek hastalığı gibi otozomal dominant hastalıkların sonraki nesillerde ortaya çıkma risklerinin yüksek olması (%50) nedeniyle bu tür bireylerin bulunduğu çiftlere PGT-M önerilmelidir.

X’e Bağlı Resesif Hastalıklar (XR)
Hemofili, Duchenne/Becker Musküler Distrofi vb. bazı hastalıklar X kromozomu üzerinde yer alan genlerden kaynaklanırlar. Bu nedenle de kalıtım modeli olarak X’e bağlı resesif olarak adlandırılırlar. X-linked resesif hastalıklar, X kromozomu ile kalıtılması nedeniyle nadir durumlar dışında sadece erkeklerde saptanır. Erkeklerde tek bir X kromozomu bulunması nedeniyle X’e bağlı resesif hastalıklar için taşıyıcılık söz konusu değildir.
Kadınlarda iki adet X kromozomu bulunduğu için sıklıkla taşıyıcılık gözlenir. Taşıyıcı kadınların erkek çocuklarında hastalığın görülme riski her gebelik için %50 iken kız çocuklarının %50’sinde taşıyıcı olma riski mevcuttur. Bu nedenle, kadında taşıyıcılık saptanan çiftlerde PGT-A işlemi için uygun tekniklerden bir tanesi seçilerek XX embriyolar seçilebilir. Ancak, taşıyıcı embriyolarında transfer edilebileceği unutulmamalıdır.
X'e bağlı resesif hastalıkların kadınlarda nadiren gözlenebildiği unutulmamalıdır. Dozaj kompanzasyonu nedeniyle kadınlarda bir X kromozomu rastgele inaktif olur ve normal şartlarda X kromozomal resesif bir hastalık için taşıyıcı olan kadında vücut ve üreme hücrelerinin yarısında normal geni taşıyan X kromozomunun, diğer yarısında ise mutant geni taşıyan X kromozomunun inaktif olması beklenir. Ancak bazen hücrelerin çoğunluğunda normal geni taşıyan X kromozomunun inaktif olması nedeniyle hastalık ortaya çıkabilir. Ayrıca, X otozomal translokasyonlarda da genetik yapının korunması amacıyla seçici olarak transloke X korunur ve diğer X kromozomu inaktif olur. Kişi translokasyonun gözlendiği X kromozomu üzerinde mutant bir gen taşıyorsa bu durum fenotipte ortaya çıkabilir.
Bu hastalıklar, nadir durumlar dışında sadece erkeklerde gözlenir. Erkeklerde tek bir X kromozomu bulunması nedeniyle X'e bağlı hastalıklar için taşıyıcılık söz konusu değildir. Hasta erkek bireylerin erkek çocuklarında hastalık veya taşıyıcılık riski bulunmaz. Ancak, kız çocuklarının tümü taşıyıcı olur. Bu nedenle, PGT uygulamasına gerek yoktur.

X’e Bağlı Dominant Hastalıklar (XD)
X'e bağlı dominant hastalıklar hem kız hem de erkek çocuklarda gözlenir. Bununla birlikte; Rett sendromu, İncontinenta pigmenti, Fokal dermal hipoplazi ve Orofaciodigital sendrom gibi bazı X'e bağlı dominant hastalıklar erkek çocuklarda meydana geldiği zaman gebelik döneminde sonlanır ve canlı doğumla bağdaşmaz. Hipofosfatemik rikets ve Frajil X gibi bazı hastalıklar ise doğum sonrası her iki cinsiyet grubunda da gözlenebilir.
Kalıtım modeli otozomal dominant kalıtıma benzer. Ancak otozomal dominant kalıtımdan farklı olarak; X’e bağlı resesif hastalıklarda olduğu gibi, hasta erkeklerin Y kromozomunu vermeleri ve hastalığın X kromozomu üzerinde kalıtılması nedeniyle erkek çocuklarda görülme riski yoktur. Hasta kadınlar da ise hastalığın kız ve erkek çocuklarda görülme riski eşit ve %50’dir.
Bu grup içerisinde yer alan hastalıklar için; ailelerde genellikle tek bir çocukta ortaya çıkması (sporadik) ve erkeklerde ölümcül, kadınlarda ise ağır seyretmesi nedeniyle PGT-M uygulamasına ihtiyaç duyulmaz. Bununla birlikte, X’e bağlı dominant hastalıklar için sağlıklı bir erkek veya kadında sadece üreme hücrelerinde mutasyonun bulunması (germline mozaisizm) gözlenebilir. Bu durum, ailede birden fazla aynı hastalığa sahip çocuk öyküsü bulunması durumunda dolaylı olarak tanımlanabilir. Bu tür ailelerde hastalığa neden olan mutasyonun bilinmesi halinde PGT-M önerilebilir.

Tek Gen Hastalıklarında PGT Uygulaması İçin Gerekli Koşullar
DNA hastalıkları için embriyolarda yapılan PGT işlemi, kan vb. örneklerden yapılan DNA testlerine göre çok daha zor olup hata payları içermektedir. Bunun en büyük nedeni, PGT işleminin embriyodan alınan tek bir blastomer veya 3-4 adet trofoektoderm örneğinden gerçekleştirilmesidir.
Tek gen hastalıklarında PGT’nin yapılabilmesi için mutlaka hastalıkla ilgili gen bölgesinin DNA testi yapılarak incelenmiş ve bu genetik bölgedeki değişimin (mutasyon) saptanmış olması gereklidir. Yapılan DNA analizlerinde sıklıkla karşılaşılan Class III (VUS - klinik etkisi bilinmeyen değişim) değişimlerin gözlendiği ailelerde tüm ekzom dizileme (WES) gibi daha geniş testlere ihtiyaç duyulabilir. Bu testler sonrasında da Class III değişim dışında başka bir değişimin bulunamaması ve bu değişimin hastalık bulguları ile uyumlu olması durumunda aile ile bu durum paylaşılmalı ve riskler konusunda bilgi verilerek ailenin yazılı onay vermesi durumunda PGT uygulanmalıdır. Ayrıca, sadece klinik olarak tanı konmuş ailelerde PGT uygulamasının yapılabilmesi teknik olarak mümkün olmadığı unutulmamalıdır.
Hastalığa neden mutasyonların belirlendiği ailelerde de PGT işleminin uygulanabilmesi için bazı şartlar mevcuttur. Bunlardan en önemlisi ön hazırlık aşaması olup PGT işleminin çok az sayıdaki hücreden elde edilecek DNA’dan yapılacak olması nedeniyle mutlaka gereklidir. Çalışmanın çok az miktardaki DNA örneğinde yapılmasından kaynaklanan sonuç alamama ve yanlış tanı gibi riskler ortaya çıkmaktadır. Bu sorunların aşılması veya en az düzeye indirilmesi için genetik laboratuvarı tarafından hem mutasyona özgü hem de incelenecek gen bölgesine yakın olan ve aileden aileye değişiklik gösteren belirteçlere özgü sentetik DNA dizileri tasarlanır. Bu tasarımlar primer üreten firmalardan temin edilerek öncelikle raporda yazan mutasyonlar doğrulanır. Hata payını azaltmak ve mutasyon çalışmasından elde edilen sonucu doğrulamak için, üretilen belirteçlerden PGT işlemi yapılacak ailede bilgi verici olanlar seçilir ve bunların tümü tek hücrede denenerek en uygun çalışma şartları belirlenir. Böylece ön hazırlık aşaması tamamlanmış olur. Bu aşamadan sonra YÜT uygulanacak merkeze bilgi verilerek hastanın tedaviye alınması sağlanır.
Ön hazırlık aşamasındaki çalışmalar nedeniyle anne, baba ve var ise hasta ve sağlıklı çocuklardan EDTA içeren tüplere alınmış kan örnekleri çifte ve/veya hastaya ait DNA raporu ile birlikte genetik tanı merkezine ulaştırılmalıdır. Bazen, taşıyıcı veya hasta anne babanın da anne babalarından ve kardeşleri ile yakın akrabalarından kan örnekleri istenmesi gerekebilir.
PGT işlemi, embriyolardan 3. gün veya 5. günde alınacak örneklerden gerçekleştirilebilir. Uzun yıllardır tek bir blastomer örneğinden uygulanmış olmakla birlikte trofoektoderm örneğinden yapılması daha yüksek sonuç elde etme oranları ve artmış güvenilirlik nedeniyle tercih edilmektedir.
PGT işlemindeki en önemli noktalardan birisi de örneklerin başka bir hücreyle kontaminasyonunun engellenmesidir. Bu nedenle, blastomer örneğini alan merkezin bu konuda deneyimli ve dikkatli olması gereklidir. Dışarıdan hücrelere bulaşacak harici bir DNA örneğinin kontaminasyona neden olmasından dolayı, yapılan tüm işlemlerde steril eldiven kullanılmasına, maske ve bone giyilmesine, kullanılan tüm besiyerlerinin ve biyopsi pipetleri ile diğer malzemelerin steril olmasına dikkat edilmelidir. Embriyolardan alınan her bir örnek ile bu embriyoların içinde bulunduğu besiyerinden negatif kontrol çalışılması amacıyla birer örnek, genetik tanı merkezi tarafından önceden hazırlanarak gönderilmiş ve içerisinde gerekli solüsyonların bulunduğu ayrı bir steril PCR tüpüne aktarılarak kapakları sıkıca kapatılır ve PGT işleminin yapılacağı merkeze uygun transfer koşullarına dikkat edilerek ulaştırılır. Bu aşamada, embriyolara ait örneklerin aktarıldığı tüplerin ve negatif kontrollerin konulduğu tüplerin numaralandırılmasına özellikle dikkat edilmelidir.
Genetik tanı merkezine ulaşan örnek tüpleri yaklaşık 45 dakikalık bir işleme tabi tutulur ve DNA elde edilir. DNA örnekleri üzerine hazırlık aşamasında tasarlanarak denenmiş özel sentetik dizilerden oluşan karışımlar ve solüsyonlar eklenerek PCR için hazırlanır. Daha sonra, PCR cihazında önceden belirlenmiş olan ısılarda işleme tabi tutularak incelenecek gen ve belirteçlere ait bölgeler çoğaltılır. Elde edilen PCR ürünü, DNA dizi ve fragman analizi yöntemleri kullanılarak DNA dizi analizi cihazında analiz edilir ve elde edilen sonuçlar değerlendirilerek tanı konur.
PGT-M uygulamasının çok az sayıda hücreden gerçekleştirilmesi nedeniyle, bir analizde kullanılabilecek en az miktardaki DNA örneği üzerinden çalışma yapılır. Bu nedenle de, bazı hücrelerde anne ve babadan gelen DNA’daki ilgili bölge PCR çalışmasında çoğaltılamaz ve buna bağlı olarak sonuç alınamayabilir (amplifikasyon başarısızlığı). Bazen de, anne veya babaya ait ilgili DNA bölgesinden sadece bir tanesi çoğaltılabilir ve bu durum allel drop-out (ADO) olarak adlandırılır.
ADO, yanlış tanıya neden olması nedeniyle PGT işlemi sırasında en çok korkulan durumdur. Bu durumun tanımlanabilmesi için aileye özgü bilgi verici belirteçlerin kullanılması bu aşamada devreye girer ve ADO varlığının tespitini sağlar. ADO saptanan örneklere ait sonuçlar, bilgi verici belirteçlerden elde edilen bilgilerle kombine edilerek nihai karar verilir ve rapor yazılır. Bu belirteçler; aynı zamanda kontaminasyon olup olmadığı konusunda da bilgi vericidir. Dahası, ilgili gen bölgesinde bir rekombinasyon olup olmadığını değerlendirmeye imkân verir. Tüm bu bilgilerin birleştirilmesiyle yine de yeterli ve güvenilir bilgiye ulaşılamazsa, o örnek değerlendirme dışı bırakılır. Değerlendirme sonrasında sağlıklı olduğu belirlenen embriyolar anne adayına transfer edilir. Gebelik oluşması durumunda, 11. haftada alınan koryonik doku örneğinde (CVS) veya 16. haftada yapılan amniyosentez işlemi sonrasında amniyon hücrelerinde PGT işleminin doğrulaması yapılmalıdır.
Tek Gen Hastalıklarında PGT Uygulamalarındaki Önemli Noktalar
1. Unutulmamalıdır ki, tüm tek gen PGT uygulamalarında %0,5 hata payı bulunmaktadır. Bunun nedeni, çalışmada herhangi bir insan hatası olmamasına rağmen yukarıda anlatılan teknik zorluklardır. Bu durum aile ile paylaşılmalı ve konu ile ilgili özel hazırlanmış bilgilendirme ve onam formları hastaya imzalatılmalıdır.
2. Özellikle; tek gen hastalıkları + HLA incelemesi yapılacak olgular ve aynı anda iki veya fazla farklı hastalık için PGT uygulanacak çiftlerde transfer edilebilecek embriyo bulmak çok daha zor olduğu ve PGT maliyetleri de yüksek olduğu için birden fazla YÜT uygulanarak embriyo biriktirme işlemi yapılması önerilmelidir. Bu tür çiftlerde, PGT işleminin tek bir çalışmada 15’e yakın sayıda embriyoda gerçekleştirilmesi hem hastaların PGT maliyetini azaltmakta hem de transfer şansını yüksek oranda arttırmaktadır.
3. Tek gen hastalıklarında PGT sonrasında zaman zaman karşılaşılan diğer bir problem de gebeliğin kromozomal hastalıklar nedeniyle kaybedilmesi veya sonlandırılmasıdır. PGT sırasında ikinci doğrulama amacıyla kullanılan bilgi verici belirteçler ayrıca ilgili genin bulunduğu kromozom için de bilgi vericidir. Bu nedenle, zaman zaman PGT raporlarında bazı embriyolar için sonuç kısmına monozomi ve trizomi gibi kromozomal hastalıklar yazılmaktadır. Bu hastalıklardan en önemlisi de Down sendromu (trizomi 21) olup sadece mutasyonun saptandığı genin 21. kromozomda olması durumunda bilgi sahibi olunabilmektedir. Bu nedenle, kromozoma ait bilgi verici belirteçlerin bulunduğu tüm tek gen PGT olgularında istenmesi halinde eş zamanlı olarak tek gen hastalığı ve 21. kromozom değerlendirmesi yapılabilir.
4. X-linked resesif hastalıklar tüp bebek aşamasında iki farklı teknik ile incelenebilmektedir. Günümüzde, X kromozomu ile kalıtılan hastalıkların DNA testleri ile embriyo aşamasında tanımlanabilmektedir. Ancak, hastalık tanısının DNA testi ile kesinleşmiş olduğu ailelerde uygulanabilmesi ve diğer tekniklere göre daha yüksek maliyete sahip olması nedeniyle pratikte daha az uygulanmaktadır. Ayrıca, FISH tekniğine göre daha gelişmiş cihaz donanımı ve bu konuda özel eğitim almış kişilere ihtiyaç durulması bu yöntemin sadece belirli merkezlerde uygulanabilmesini mümkün kılmaktadır. PGT uygulamasını yapacak merkezin bu teknolojiye sahip olmadığı veya hastalık tanısının sadece klinik olarak konabildiği aileler ile ekonomik olarak bu uygulamayı karşılaması mümkün olmayan çiftlerde FISH, aCGH veya NGS tekniği kullanılmaktadır. Ülkemizdeki yasalara göre cinsiyet tayini yasaklanmış olmakla birlikte bu tür X'e bağlı kalıtılan hastalıklar için embriyo aşamasında cinsiyet seçimine izin verilmiştir.
5. Bazı ailelerde ikinci doğrulama testi olan bağlantı analizinde kullanılan bilgi verici belirteçler o aileye özgü olarak bilgi verici veya istenilen yeterlilikte olmayabilir. Bu durum; özellikle dominant hastalıklarda hata payının yükselmesine neden olur.
6. Nörofibromatozis gibi otozomal dominant bir hastalığın gözlendiği bazı çiftlerde mutasyonun de novo olarak ilk o kişide oluşması nedeniyle ailede başka etkilenen birey bulunmaz. Bu nedenle de, PGT hazırlık aşamasında bağlantı analizi için gerekli olan bilgi verici belirteçler belirlenemeyebilir. Hasta kişi erkek ise, ikinci bir seçenek olarak baba adayından semen örneği alınır ve haploid sperm hücrelerinde tek tek çalışma yapılarak bilgi vericibelirteçlerden bağlantı analizi için kalıtım bilgisi elde edilmeye çalışılır. Aksi halde, PGT hazırlık aşamasında sadece mutasyon için ön çalışma yapılabilir. Bu tür çiftlerde, bağlantı analizi için gerekli olan bilgiye, embriyo çalışmasından elde edilen sonuçlardan yola çıkılarak ulaşılabilir. Embriyo sayısının yeterli bağlantı analizi bilgisine ulaşamayacak kadar az olması durumunda ve mutasyonun de novo olması durumunda bu da mümkün olmayabilir.
7. Sağlıklı bir çiftte Nörofibromatozis gibi otozomal dominant bir hastalığa sahip çocuğun bulunması durumunda ise, mutasyonun de novo mu olduğu yoksa üreme hücrelerindeki mozaisizmden mi kaynaklandığı bilenememektedir. Mozaisizm riskinden dolayı PGT bu ailelere önerilmektedir. Benzer olarak, mozaisizmin erkekte olma ihtimali düşünülerek baba adayından semen örneği alınır ve sperm hücrelerinde tek tek çalışma yapılarak germline mozaisizm olup olmadığı bilgisine ulaşmaya çalışılır. Mozaisizm bilgisi bu çalışmadan elde edilemez ise embriyo çalışmasından elde edilen sonuçlardan yola çıkılarak anlaşılmaya çalışılır. Embriyo sayısının yeterli bilgi verecek sayıda olmaması durumunda ve mutasyonun de novo olması durumunda bu da mümkün olmayabilir.
8. Bazı istisnalar dışında; DMD/BMD, Alfa Talasemi gibi bazen gen duplikasyonları ve büyük gen delesyonlarından kaynaklanan hastalıklarda embriyolarda mutasyon analizi yapmak mümkün olmayabilir. PGT işlemi sadece bağlantı analizi ile gerçekleştirilebilir. PGT uygulamasının yapılabilmesi için de ailede aynı mutasyon için hasta, taşıyıcı ya da normal bir bireyin bulunması gereklidir.
9. Bazı ailelerde ikinci doğrulama testi olan bağlantı analizi için gerekli olan belirteçler o aileye özgü olarak bilgi verici veya istenilen yeterlilikte olmayabilir. Bu durum; özellikle dominant hastalıklarda hata payının yükselmesine neden olur.
10. Talasemi gibi tedavi amaçlı kan transfüzyonu uygulanan (3 ay içerisinde) veya kemik iliği nakli yapılan hastalıklarda, hasta bireylerde bu işlemlerden birisi daha önce yapılmış ise ön hazırlık aşamasında kullanılmak üzere gerekli olan doğru örnek tipi steril olarak alınmış bukkal swap (yanak içi mukoza) örneğidir. Örnekler 2-3 adet olacak şekilde jelsiz (kuru) bukkal swap şeklinde alınmalı ve gönderilmelidir.
11. Frajil X, Huntington, Myotonik Distrofi gibi trinükleotid tekrar hastalıklarında, embriyolarda mutasyon analizi yapmak teknik olarak mümkün olmadığı durumlarda PGT işlemini sadece bağlantı analizi ile gerçekleştirilmek gerekebilir. Bu durumda da, PGT uygulamasının yapılabilmesi için ailede hasta ya da normal ikinci bireyin bulunması gereklidir. Aksi halde PGT yapmak mümkün değildir. Sadece, X’e bağlı hastalıklarda FISH veya aCGH/NGS tekniklerinden birisi kullanılarak XX embriyolar seçilebilir.
12. Frajil X taşıyıcısı kadınlarda ve erkeklerde FMR1 geni mutasyon analiz raporları iyi değerlendirilmeli ve tekrar sayısına göre PGT gerekliliği konusunda karar verilmelidir. Normal bireylerde FMR1 genindeki CGG tekrar sayıları 5-44 arasında değişkenlik gösterir. Gri bölge (intermediate) olarak adlandırılan 45-54 arasındaki tekrar sayılarına sahip bireylerin çocuklarında hastalık gözlenme riski olmadığı için PGT uygulanmasına gerek yoktur. Bu kişilerin çocuklarında ise tekrar sayıları artarak premutasyon olarak adlandırılan 55-200 sayısına çıkabilir. Bu nedenle, kendi çocukları için risk olmamakla beraber bir sonraki kuşakta risk ortaya çıkabilir. Premutasyon taşıyıcı kadınlar Frajil X sendromlu çocuk açısından riske sahip oldukları için PGT önerilmelidir. PGT işlemi daha öncede belirtildiği gibi sadece gerekli ön hazırlık koşullarına sahip çiftlerde bağlantı analizi ile uygulanabilir. Premutasyon taşıyıcı erkeklerde ise kadınların aksine çok küçük tekrar artışları meydana geldiği için kız çocuklarının tümü premutasyon taşıyıcı olurlar. Erkek çocuklarına Y kromozomu aktarıldığı için hastalık riski yoktur.
13. Huntington hastalığı (HD), Frajil X gibi bir trinükleotid tekrar hastalığı olup HTT genindeki CAG trinükleotid tekrar sayısı artışlarından kaynaklanır. Hastalık geç dönem başlangıçlı olduğu için taşıyıcı sağlıklı veya hasta (belirtiler mevcut veya değil) bireyler PGT işlemine ihtiyaç duyabilirler. Normal bireylerde HTT genindeki CAG tekrar sayıları 27 veya daha az sayıdadır. Orta büyüklükte (intermediate) tekrar sayıları olarak kabul edilen ve hastalık riskinin bulunmadığı kişiler ise 27-35 arasındaki tekrar sayılarına sahiptirler. Bu kişilerin çocuklarında tekrar sayısının artmasına bağlı HD hastalığı görülme riski arttığı için PGT önerilmelidir. Tekrar sayısı 36-39 arasında olan kişilerde her zaman hastalık belirtileri ortaya çıkmamakla birlikte bu alleli çocuklarına aktarma riskleri %50 olduğu için PGT uygulanmalıdır. Yine, hastalık bulgularının yaşam içerisinde mutlaka gözlendiği 40 ve üzerindeki tekrar sayılarının bulunduğu bireylerde de hasta çocuk riski %50 olduğu için mutlaka PGT önerilmelidir. PGT işlemi daha önce belirtildiği gibi sadece gerekli ön hazırlık koşullarına sahip çiftlerde uygulanabilir.
14. Myotonik Distrofi (MD) diğer sık trinükleotid tekrar hastalığı olup DMPK genindeki CTG trinükleotid tekrar sayısı artışlarından kaynaklanır. Normal bireylerde DMPK genindeki CTG tekrar sayıları 5-34 arasındadır. Premutasyon olarak kabul edilen ve hastalık riskinin bulunmadığı kişiler 35-49 arasındaki tekrar sayılarına sahiptirler Bu kişilerin çocuklarında tekrar sayıları artarak hastalık gözlenebileceği için PGT önerilmelidir. Yine, hastalık bulgularının gözlendiği 50 ve üzerindeki tekrar sayılarının bulunduğu bireylerde de hasta çocuk doğma riskinin %50 olması nedeniyle mutlaka PGT önerilmelidir. PGT işlemi daha önce belirtildiği gibi sadece gerekli ön hazırlık koşullarına sahip çiftlerde uygulanabilir.
15. Bazı tek gen hastalıkları PGT uygulamalarında eş zamanlı olarak 24 kromozom incelemesi de istenmektedir. Bu tür çiftlerde her iki incelemenin de yapılabilmesi için 5. gün trofoektoderm biyopsisi uygulaması PGT sonuçlarının güvenilirliği açısından gereklidir. Ancak iki farklı teknik ile gerçekleştirilebilecek olan bu hastalardaki PGT için birkaç farklı yol izlenebilir. Her embriyodan alınan iki farklı trofoektoderm veya tek bir örnek alınıp daha sonra iki parçaya ayrılan örnekler 1A, 1B gibi ayrı kodlar ile tüplere aktarılarak gönderilebilir. Her bir embriyoya ait ilk örnek (1A) PGT-M uygulamasında kullanılır. Tüm embriyoların analizi sonrasında, tek gen hastalığı açısından sağlıklı olduğu belirlenen embriyolara ait ikinci örnekler (1B) tüm genom çoğaltması (WGA) yapılarak aCGH veya NGS çalışmasına alınarak 24 kromozom incelemesi gerçekleştirilebilir. Bu sayede hem güvenilir sonuçlar elde edilebilir hem de sadece DNA hastalığı açısından sağlıklı olan embriyolara PGT-A uygulandığı için hasta açısından maliyetler en düşük düzeyde kalır. Bununla birlikte, çift biyopsi veya tek biyopsi alınıp iki parçaya ayrılması çok tercih edilen bir işlem değildir. Daha sık kullanılan yöntem ise, tek bir trofoektoderm örneği alınması ve laboratuvara gönderilmesi olup farklı bir yol izlenerek PGT-M ve PGT-A analizlerinin gerçekleştirilmesidir. Bu tür örneklerde, PGT-M için gerekli olmamasına rağmen PGT-A için zorunlu olan WGA işleminin ilk aşamada yapılması zorunludur. Elde edilen WGA ürününün yaklaşık dörtte biri kullanılarak önce tek gen hastalığı için PGT yapılır. Çalışma sonrası sağlıklı olduğu saptanan embriyolarda yine WGA ürünün bir kısmı daha kullanılarak PGT-A gerçekleştirilebilir. Bu tür uygulamalarda, ön hazırlık aşamasının WGA ürününden tek gen tanımlaması yapılacağı için daha farklı yapılması gereklidir. Bu nedenle de, embriyolarda PGT-M ve PGT-A çalışmasının her ikisinin de yapılmasının istendiği daha ön hazırlık aşaması başlamadan önce genetik tanı merkezine belirtilmelidir. Tek biyopsi örneğinden iki farklı PGT işleminin uygulanması pratik olmakla birlikte WGA sonrasında PGT-M yapılması nedeniyle çalışma kalitesi zaman zaman biyopsi örneklerinin DNA veya WGA ürününün kalitesinden etkilenebildiği unutulmamalıdır. Bu da, bazı embriyolardaki sonuçları yorumlamakta zorluklara neden olabilir.
TEK GEN HASTALIKLARI + DOKU TİPLEMESİ (HLA)
Doğumdan sonra klinik belirtileri ortaya çıkan genetik hastalıklar içerisinde yer alan Talasemi, Orak Hücre Anemisi gibi kan hastalıkları ile bağışıklık sistemi hastalıkları ilaçlar ile kesin olarak tedavi edilememektedir. Uygun doku tipine sahip kemik iliği bulunması durumunda ise nakil ile kesin tedavi sağlanabilmektedir. Ayrıca, hematolojik ve bağışıklık sistemi hastalıklarına sahip çocukları bulunan ailelerde her gebelikte hastalığın tekrarlama riskleri mevcuttur. Bu risk, otozomal resesif hastalıklarda %25, otozomal dominant hastalıklar için %50 ve X-linked resesif hastalıklar da sadece erkek çocuklarda olmak üzere %50'dir.
Bu ailelerde; öncelikle, hasta çocuk ile anne, baba, kardeş ve 1. derece akrabaların HLA tipleri karşılaştırılarak uygun verici araştırılmakla birlikte HLA bölgesinin çok polimorfik bir yapıda olması nedeniyle genellikle olumlu sonuç alınamaz. Günümüzde, PGT yöntemi ile sağlıklı embriyoların saptanmasının yanı sıra HLA (doku) tiplemesi işlemi de aynı anda uygulanabilmekte ve embriyoların doku tipi belirlenebilmektedir.
PGT Uygulaması İçin Gerekli Koşullar
Embriyolarda tek gen hastalığı ile birlikte HLA tiplemesinin de yapılacak olması, sadece tek gen hastalığı bakılacak olgulara göre ön hazırlık aşaması açısından değişiklikler ve ilaveler gerektirir. Talasemi gibi kan transfüzyonu uygulanan hastalıklarda hasta çocuktan alınacak örnek tipinin EDTA’lı kan örneği yerine bukkal swap olması zorunludur. Aksi halde, hasta çocuğun HLA genotipini hazırlık aşamasında doğru olarak belirlemek teknik olarak mümkün değildir. Benzer olarak kemik iliği nakli yapılmış bireylerden de aynı şekilde örnek tipi gereklidir.
Ön hazırlık aşamasındaki diğer bir farklılık ise uygun HLA belirteçlerinin belirlenmesi ve çalışmaya dahil edilmesidir. HLA bölgesinin genetik olarak polimorfik ve büyük bir yapıda olması nedeniyle doğru tanımlama için bölge içerisinde en alt, orta ve üst bölgelerin her birisi için en az 3 adet olacak şekilde STR seçilmesi önemlidir.
Yapılacak işleme HLA belirteçlerinin de eklenmesi nedeniyle ön hazırlık aşamasında çalışma optimizasyonu yapmak ve embriyoda da uygulamak teknik olarak daha zordur.
Diğer bir önemli nokta, hem tek gen hastalığından etkilenmemiş hem de doku tipi hasta çocuk ile aynı olan embriyoların tanımlanmaya çalışılmasından dolayı bu iki şartı aynı anda taşıyan embriyo bulmadaki zorluktur. Bu oran, sadece Talasemi için yapılan PGT çalışmaları ile karşılaştırıldığında Talasemi + HLA olgularında çok daha düşük olup yaklaşık olarak %10-15’dir.
DİĞER
DOKU TİPLEMESİ (HLA)
Çocukluk çağında sık gözlenen hematolojik kanserler, uygun doku tipine sahip kemik iliği bulunması durumunda etkin bir şekilde tedavi edilebilmektedir. Öncelikle, hasta çocuk ile anne, baba, kardeş ve 1. derece akrabaların HLA tipleri karşılaştırılarak uygun verici araştırılmakla birlikte HLA bölgesinin çok polimorfik olması nedeniyle genellikle olumlu sonuç alınamaz.
Lösemi gibi sonradan kazanılmış hematolojik hastalıkların gözlendiği ailelerde, gebelik öncesi genetik tanı yöntemi ile embriyolarda HLA (doku) tiplemesi işlemi yapılabilmektedir. Preimplantasyon genetik tanı (PGT), bu tür hastalıkların tedavisindeki güçlükler ve yüksek tedavi maliyetleri düşünüldüğünde çok önemli bir tekniktir.
PGT Uygulaması İçin Gerekli Koşullar
Bu hastalarda PGT uygulaması için tüm koşullar tek gen hastalığı + HLA uygulaması ile aynı olup tek farkı uygun HLA tipine sahip embriyo bulma şansının daha yüksek olmasıdır.
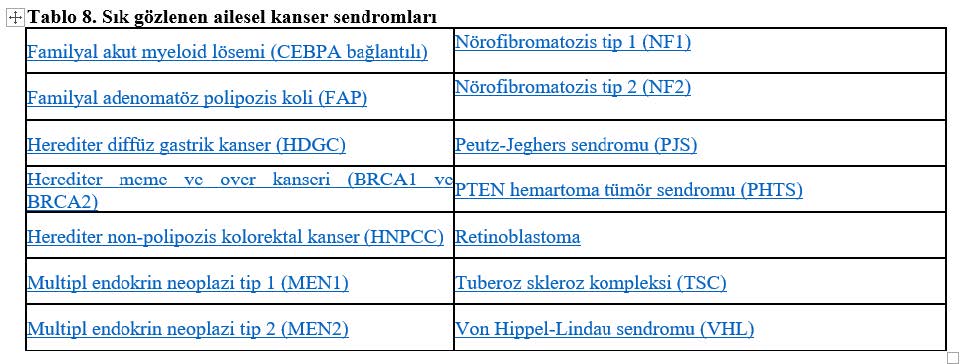
AİLESEL KANSER SENDROMLARI
Genellikle tümör süpressör genlerde (TSG) meydana gelen mutasyonlar başlangıçta germline olarak sperm veya yumurta hücresinde meydana gelerek doğacak olan kişiye kalıtılır. Yaşam içerisinde aynı genin diğer allellinde meydana gelen ikinci bir mutasyon sonrasında kanser oluşumu gözlenir.
Her iki allelde de mutasyon oluşması sonrasında TSG inaktif olarak hücre proliferasyonu üzerindeki süpressör etkisini kaybeder. Daha nadir olmakla birlikte, anne ve babadan normal olarak kalıtılan bir TSG'de yaşam içerisinde aynı hücrede olmak kaydı ile iki mutasyon gözlenebilir. Ancak, mutasyonlardan birisini kalıtsal olarak ailesinden alarak tüm hücrelerinde taşıyan bir kişide etkilenen hücre sayısının fazlalığı nedeniyle ikinci bir mutasyonun oluşması ve buna bağlı olarak kanser gelişimi çok daha olasıdır.
Ailesel meme ve over kanserine neden olan BRCA1 ve BRCA2 genleri ailesel kanser sendromları için en iyi örneklerden bir tanesidir. BRCA1 mutasyonunu taşıyan kadınlarda meme kanseri ve over kanseri gelişme riski sırasıyla %85 ve %45’dir. BRCA2 mutasyonlarında ise BRCA1 mutasyonları ile benzer olarak artmış meme kanseri riski gözlenirken over kanseri için bu risk %27’dir.
BRCA1, BRCA2, P53, MLH1, MSH2, APC gibi bazı gen bölgelerindeki mutasyon kanser yatkınlığına ve ailesel kanser sendromlarının gözlenmesine neden olabilir. Bu mutasyonların üreme hücreleri yoluyla sonraki nesillere aktarılma riski %50 olduğu için bu tür çiftlere PGT önerilebilmektedir.
Günümüzde bu tür sendromlara neden olan genetik bölgeler incelenerek risk altındaki kişiler belirlenebilmektedir. Dahası, bu tür genetik bölgelerde mutasyonların saptandığı kişilerin birinci derece akrabalarında ve çocuklarında artmış kanser riski nedeniyle PGT uygulaması ile bu klinik tablonun önüne geçilebilmektedir.
PGT Uygulaması İçin Gerekli Koşullar
Bu genlerin etki açısından resesif kalıtım modeline uymasına rağmen (P53 haricinde) mutasyonu alan embriyoların etkilenmiş kabul edilmesinden dolayı PGT işlemi otozomal dominant tek gen hastalıklarındaki PGT uygulama koşullarına göre yapılmalıdır.
Rh Uyuşmazlığı
Rh uyuşmazlığı, gebeliklerde sık karşılaşılan bir durum olup anne adayının Rh(-) baba adayının ise Rh(+) olduğu durumlarda söz konusudur. Bu durumda, bebeğin Rh(+) olma ihtimali söz konusu olduğu için gerekli önlemlerin alınması gereklidir.
Rh uyuşmazlığındaki en büyük teknik sorun, Rh(+) bir baba adayının anne ve babasından aldığı Rh tipinin biyokimyasal olarak belirlenmesinin çok zor olması ve dünyada birkaç merkezde yapılabilmesidir. Bu nedenle, Rh(+) olduğu bilinen bir kişinin Rh genotipinin (+/+) veya (+/-) olup olmadığı önceden tanımlanabilmesi için kan örneği bu çalışmayı yapan bir merkeze gönderilmelidir. Diğer bir seçenek de, baba adayından semen örneği almak ve haploid sperm hücrelerini tek tek çalışarak Rh(-) sperm hücrelerinin var olup olmadığını belirlemektir. Özellikle; (+/+) genotipe sahip olduğu saptanan bir babanın varlığında anne Rh(-) ise çiftlerin tüm gebeliklerinde bebekler Rh(+) genotipine sahip olacağı için PGT uygulaması yapılmamalıdır.
Günümüzde, gerekli önlemlerin alınmadığı veya alınan önlemlere rağmen sorunların gözlendiği çiftlerin yeni gebeliklerinde tüp bebek yöntemi uygulanması durumunda embriyo aşamasında bebeğin Rh tipi belirlenebilmekte ve Rh(-) embriyolar seçilebilmektedir.
PGT Uygulaması İçin Gerekli Koşullar
Bu tür çiftlerde, PGT öncesinde Rh genotiplemesiyle ve haploid sperm genomu çalışarak babanın Rh genotipinin (+/-) olduğu belirlenir. Sperm çalışmasında, Rh genotipinin belirlenmesinde kullanılan RHD ve RHCE genleriyle beraber bu genlerin sentromerik ve telomerik yakın komşuluğundaki STR markörleri de çalışılarak babanın Rh(-) ve Rh(+) haplotipleri tespit edilmektedir. Sperm çalışmasına babayla beraber anne DNA’sı da ilave edilerek her iki ebeveynin de STR haplotipleri tespit edilmiş olur. Bu ön hazırlık çalışmasında Rh(-) ve Rh(+) alleller ile beraber aktarılan STR haplotipleri belirlenmiş olur.
Daha sonra, çift IVF tedavisine alınmakta ve embriyolardan alınan örneklerde Rh genotipine bakılmaktadır. Yapılan işlemde, (-/-) genotipe sahip embriyolar seçilmektedir. Diğer bir deyişle analizler, varsa Rh(-) embriyoların tanımlanması amacıyla yapılmaktadır.

Referanslar
1. Ao A, Ray P, Harper J, Lesko J, Paraschos T, Atkinson G, Soussis I, Taylor D, Handyside A, Hughes M, Winston RM. Clinical experience with preimplantation genetic diagnosis of cystic fibrosis (delta F508). Prenat Diagn. 1996 Feb;16(2):137-42.2. Munné S, Weier HU. Simultaneous enumeration of chromosomes 13, 18, 21, X, and Y in interphase cells for preimplantation genetic diagnosis of aneuploidy. Cytogenet Cell Genet. 1996;75(4):263-70. 3. De Vos A, Sermon K, Van de Velde H, Joris H, Vandervorst M, Lissens W, De Paepe A, Liebaers I, Van Steirteghem A. Two pregnancies after preimplantation genetic diagnosis for osteogenesis imperfecta type I and type IV. Hum Genet. 2000 Jun;106(6):605-13.
4. Ray PF, Gigarel N, Bonnefont JP, Attié T, Hamamah S, Frydman N, Vekemans M, Frydman R, Munnich A. First specific preimplantation genetic diagnosis for ornithine transcarbamylase deficiency. Prenat Diagn. 2000 Dec;20(13):1048-54.
5. Sermon K, Seneca S, De Rycke M, Goossens V, Van de Velde H, De Vos A, Platteau P, Lissens W, Van Steirteghem A, Liebaers I. PGD in the lab for triplet repeat diseases - myotonic dystrophy, Huntington's disease and Fragile-X syndromeMol Cell Endocrinol. 2001 Oct; 22;183 Suppl 1:S77-85.
6. Kuliev A and Verlinsky Y. Current features of preimplantation genetic diagnosis. Reprod Biomed Online 2002; 5, 294–299.
7. Results of preimplantation genetic diagnosis in patients with Klinefelter's syndrome. Kahraman S, Findikli N, Berkil H, Bakircioglu E, Donmez E, Sertyel S, Biricik A. Reprod Biomed Online. 2003 Oct;7(3):346-52.
8. Embryo development characteristics in Robertsonian and reciprocal translocations: a comparison of results with non-translocation cases. Findikli N, Kahraman S, Kumtepe Y, Donmez E, Biricik A, Sertyel S, Berkil H, Melil S. Reprod Biomed Online. 2003 Nov;7(5):563-71.
9. Kahraman S, Benkhalifa M, Donmez E, Biricik A, Sertyel S, Findikli N, Berkil H. The results of aneuploidy screening in 276 couples undergoing assisted reproductive techniques. Prenat Diagn. 2004 Apr;24(4):307-11.
10. Fiorentino F, Biricik A, Karadayi H, Berkil H, Karlikaya G, Sertyel S, Podini D, Baldi M, Magli MC, Gianaroli L, Kahraman S. Mol Hum Reprod. Development and clinical application of a strategy for preimplantation genetic diagnosis of single gene disorders combined with HLA matching. 2004 Jun;10(6):445-60.
11. Seeho SK, Burton G, Leigh D, Marshall JT, Persson JW, Morris JM. The role of preimplantation genetic diagnosis in the management of severe rhesus alloimmunization: first unaffected pregnancy: case report. Hum Reprod. 2005 Mar;20(3):697-701.
12. Wilton L, Thornhill A, Traeger-Synodinos J, Sermon KD, Harper JC. The causes of misdiagnosis and adverse outcomes in PGD. Hum Reprod. 2009 May;24(5):1221-8.
13. Johnson DS, Cinnioglu C, Ross R, Filby A, Gemelos G, Hill M, Ryan A,Smotrich D, Rabinowitz M, Murray MJ. Comprehensive Analysis of Karyotypic Mosaicism between Trophectoderm and Inner Cell Mass. Mol. Hum.Reprod. 2010 Dec;16(12):944-9.
14. Johnson DS, Gemelos G, Baner J, Ryan A, Cinnioglu C, Banjevic M, Ross R, Alper M, Barrett B, Frederick J, Potter D, Behr B, Rabinowitz M. Preclinical Validation of a Microarray Method for Full Molecular Karyotyping of Blastomeres in a 24-hour Protocol. Hum Reprod. 2010 Apr;25(4):1066-75.
15. Gutiérrez-Mateo C1, Colls P, Sánchez-García J, Escudero T, Prates R, Ketterson K, Wells D, Munné S. Validation of microarray comparative genomic hybridization for comprehensive chromosome analysis of embryos. 2011 Mar 1; Fertil Steril, 95:953–8.
16. Alfarawati S, Fragouli E, Colls P, Wells D. First births after preimplantation genetic diagnosis of structural chromosome abnormalities using comparative genomic hybridization and microarray analysis. Hum Reprod. 2011 Jun;26(6):1560-74.
17. Fiorentino F, Biricik A, Bono S, Spizzichino L, Cotroneo E, Cottone G, Kokocinski F, Michel CE. Development and validation of a next-generation sequencing-based protocol for 24-chromosome aneuploidy screening of embryos. Fertil Steril. 2014 May;101(5):1375-82.
18. Francesco Fiorentino, Sara Bono, Anil Biricik, Andrea Nuccitelli, Ettore Cotroneo, Giuliano Cottone, Felix Kokocinski, Claude-Edouard Michel, Maria Giulia Minasi, and Ermanno Greco. Application of next-generation sequencing technology for comprehensive aneuploidy screening of blastocysts in clinical preimplantation genetic screening cycles. Human Reproduction, 2014; Vol.29, No.12 pp. 2802–2813,.
19. Wells D, Kaur K, Grifo J, Glassner M, Taylor JC, Fragouli E, Munne S. Clinical utilisation of a rapid low-pass whole genome sequencing technique for the diagnosis of aneuploidy in human embryos prior to implantation. J Med Genet. 2014 Aug;51(8):553-62.
20. Tan Y, Yin X, Zhang S, Jiang H, Tan K, Li J, Xiong B, Gong F, Zhang C, Pan X, Chen F, Chen S, Gong C, Lu C, Luo K, Gu Y, Zhang X, Wang W, Xu X, Vajta G, Bolund L, Yang H, Lu G, Du Y, Lin G. Gigascience. Clinical outcome of preimplantation genetic diagnosis and screening using next generation sequencing. 2014 Dec 4;3(1):30.
21. Haiyan Zheng, Hua Jin, Lian Liu, Jianqiao Liu, and Wei-Hua Wang. Application of next-generation sequencing for 24-chromosome aneuploidy screening of human preimplantation embryos. Mol Cytogenet. 2015; 8: 38.
22. Kung A, Munné S, Bankowski B, Coates A, Wells D. Validation of next-generation sequencing for comprehensive chromosome screening of embryos. Reprod Biomed Online. 2015 Dec;31(6):760-9.
23. https://www.illumina.com/products/by-type/clinical-research-products/veriseq-pgs.html
24. https://www.thermofisher.com/tr/en/home/life-science/sequencing/dna-sequencing/preimplantation-geneticscreening.html
25. https://www.ogt.com/products/936_cytosure_embryo_screen_array
26. https://www.illumina.com/clinical/reproductive-genetic-health.html